CloudOS Meets nf-core: Standardising Cloud-Native Bioinformatics Pipelines

Bioinformaticians in the pursuit of best practices
The democratisation of Next Generation Sequencing technologies has made omics data more available than ever before. Bioinformaticians are now confronted with tackling the NGS data deluge in order to deliver actionable insights. Transforming raw data into impactful insights requires bioinformaticians to process omics data with bioinformatics pipelines. To date, however, no validated standards have been widely established by the bioinformatics community. The push towards standardised bioinformatics pipelines is critical for enhancing reproducibility and efficiency in research.
The integration of standardised bioinformatics pipelines in research can significantly enhance the reliability of results.
As with omics data, the landscape of bioinformatic tools and software is also evolving at a rapid pace, making it hard for bioinformaticians to keep up with the latest releases. However, some workflows are considered best practice in the field, as is the case with the GATK best practices workflow for standardising secondary NGS data analysis as defined by the Broad Institute, and Illumina’s DRAGEN the accelerated version of BWA/GATK that I extensively discussed in my previous blog post.
Although best practices are recommended for use of the GATK workflow, issues arise when the implementation is left in the hands of individual researchers, especially when it is customised to run in local environments, ultimately preventing interoperability, a central pillar of the FAIR (findable, accessible, interoperable, reusable) principle.
Utilising standardised bioinformatics pipelines ensures that researchers can replicate their findings across different studies.
As the demand for consistency grows, adopting standardised bioinformatics pipelines will help researchers ensure that their analyses are both robust and repeatable.
The development of scientific workflow management systems stems from reproducibility, standardisation and portability issues in bioinformatics
By adopting standardised bioinformatics pipelines, researchers can effectively address the increasing demands for reproducibility in their analyses.
Standardised bioinformatics pipelines can also streamline workflows and facilitate collaboration among scientists.
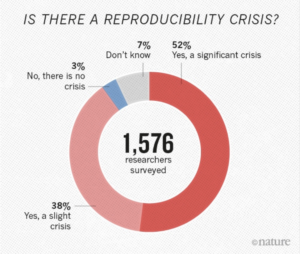
There is growing alarm about results that cannot be reproduced, especially in the field of bioinformatics. About 52% of researchers agree that there is a reproducibility crisis (read about how some researchers tackle reproducibility within their own laboratory). Reproducibility, in addition to the rise of standardisation, portability, and data governance issues in bioinformatics, has led to the organic development of powerful scientific workflow management systems which allow users to build pipelines and improve portability by abstracting the computing infrastructure from the pipeline logic.
Workflow management systems capture the exact methodology that bioinformaticians have followed for a specific in silico experiment, thereby greatly improving the reproducibility of computational analyses.
A widely adopted workflow management system, Nextflow, is an open-source container-based programming framework which allows workflows to run more efficiently. More than half of all big international pharmas involved in bioinformatic analysis have adopted Nextflow, as it has been demonstrated to be superior in terms of scalability and data handling, and more numerically stable than other open-source workflow management.
Nextflow is based on the dataflow programming model which simplifies and streamlines writing complex distributed pipeline. Parallelisation is implicitly defined by the processes input and output declarations, thereby producing parallel applications that can easily be scaled up or down, depending on real-time compute requirements. The automation that is inherent to Nextflow minimises human error and increases reproducibility among different compute environments.
A community-driven initiative to implement best practices for Nextflow pipeline development
We encourage developers to contribute to the creation of standardised bioinformatics pipelines that can benefit the entire research community.
Through the use of standardised bioinformatics pipelines, Lifebit aims to enhance data accessibility and reproducibility.
Standardised bioinformatics pipelines ensure that researchers can focus on their scientific questions without getting bogged down by technical details.
Running standardised bioinformatics pipelines via CloudOS enhances the overall efficiency of bioinformatics workflows.

With standardised bioinformatics pipelines, organizations can leverage existing best practices across projects.
In late 2017, the nf-core framework was created to remove existing silos amongst bioinformaticians working on their own isolated copies of workflows built with Nextflow. nf-core’s ultimate goal is to tear down barriers in order to work together as a community to deliver best practice, peer-reviewed pipelines that can be used by anyone, whether it be individual researchers, developers or research facilities. nf-core pipelines are easy to use (i.e. can be run with a single command!), are bundled with containers with all of the required software dependencies (Docker or Singularity), have continuous integration testing, stable release tags, and provide extensive documentation.
The ways Lifebit has been contributing to Nextflow best practices & nf-core
Last year, Lifebit contributed to the nf-core community initiative by developing our DeepVariant pipeline adhering to nf-core guidelines. If you’re curious about what it takes to develop a best practice nf-core pipeline, check out Lifebit Bioinformatician, Phil Palmer’s account of his experience whilst making a pipeline nf-core compliant.
One of Lifebit’s core missions is to make all data, tools and computational resources needed to run reproducible bioinformatics analyses available in one place. Therefore, to facilitate the use of gold-standard Nextflow pipelines, we have made available all of the stable nf-core pipelines on the CloudOS platform, which will allow CloudOS users to easily access nf-core approved pipelines within their own CloudOS environment.
The benefits of running nf-core bioinformatics pipelines through CloudOS in your own AWS or other cloud
Implementing standardised bioinformatics pipelines can lead to significant time savings in project development.
Researchers who use standardised bioinformatics pipelines find it easier to share their results with peers.
The future of bioinformatics relies on the widespread adoption of standardised bioinformatics pipelines.
We invite feedback on how standardised bioinformatics pipelines can be further improved in collaborative efforts.
Why run nf-core stable release pipelines over CloudOS rather than the command line interface, you ask? At Lifebit, we always prioritise CloudOS features that reinforce the concepts of standardisation, reproducibility and auditability, thereby ensuring that all stages of bioinformatics analysis are FAIR. By running nf-core stable release bioinformatics pipelines in CloudOS, you will be able to:
- Run nf-core stable release pipelines through a graphical user interface (GUI), where you benefit from both job deployment and scheduling
- Manage all your data, environment and pipelines in one place
- Scale – you have access to a near-infinite compute infrastructure
- Monitor jobs in real-time and easily visualise results
- Painlessly collaborate with the rest of your team through 1-click cloning and sharing of any analysis
- Reduce costs of running nf-core stable release pipelines with AWS spot instances
- Above all, have analysis versioning with the only GitHub-like system that can version all the elements of analysis (data, pipeline, resources used, costs, owner, duration, timestamps etc.) – instead of just having only the nf-core pipeline versioning
Stay tuned as we will be continuously growing our pipeline catalogue on CloudOS as more stable releases of Nextflow-based pipelines are curated and released via the nf-core initiative.
A special thanks to the nf-core team for their help & suggestions with this article!
We would like to know what you think! Please fill out the following form or contact us at hello@lifebit.ai. We welcome your comments and suggestions!