

Share:

The beginning of the 21st century has been shaped by the fast growing amount of biological data which is produced by sequencing technologies and, in particular, by the high throughput next generation sequencing (NGS). Genetic variations in human genomes, especially the very common single nucleotide polymorphisms ( SNPs ), are correlated with many diseases 1, associated with individuality 2 and relevant to many other fields, such as nutrition 3. In the last few years, interest in genomic variations and their effects on phenotype and impact on diseases has been growing. The first step to harness the genotype-phenotype connection is to be able, given an individual, to identify the variations in his/her genome.
Variant Calling tools identify genetic variations of newly sequenced genomes by comparing to the corresponding reference genome. In practice, the output of NGS technologies (reads) are mapped to the reference genome and variations are identified. In the last years, many variant callers have been released in order to obtain accurate and fast results. In December 2017, the team at Google Brain joined the effort by releasing an open source, deep learning based variant caller: DeepVariant. DeepVariant outperforms its competitors by accuracy – it even won the accuracy award at the precision FDA Truth Challenge.
As we started using DeepVariant, we realised that running the binaries provided by Google over our datasets was not efficient enough. Parallelisation over multiple files is not handled by default, input files need some preprocessing since not all of them are compatible with DeepVariant, and manually running the binaries or, writing some temporary script on top of them, was not sustainable. Moreover, we needed a way to seamlessly run DeepVariant over cloud in the easiest way possible. That’s when we decided to write a Nextflow-based pipeline and use it on CloudOS to solve our problems.
DeepVariant as a Nextflow-based pipeline enables users to run DeepVariant in an easy, fast and reproducible manner that ensures full control over configurations. It supports the user to start running the pipeline correctly and it allows running of multiple variant calling processes in parallel to maximise efficiency. Every step is bundled and computed in a Docker container. Full support to easily run and scale analyses with DeepVariant over AWS and Azure cloud is provided by CloudOS.
DeepVariant: an overview
DeepVariant performs variant calling through image recognition. Images representing the mapping of the reads to the reference genome are produced using BAM files. The picture below, taken from the official DeepVariant google blog post, shows examples of such images. These images are then used by the trained machine learning model to identify the variants.
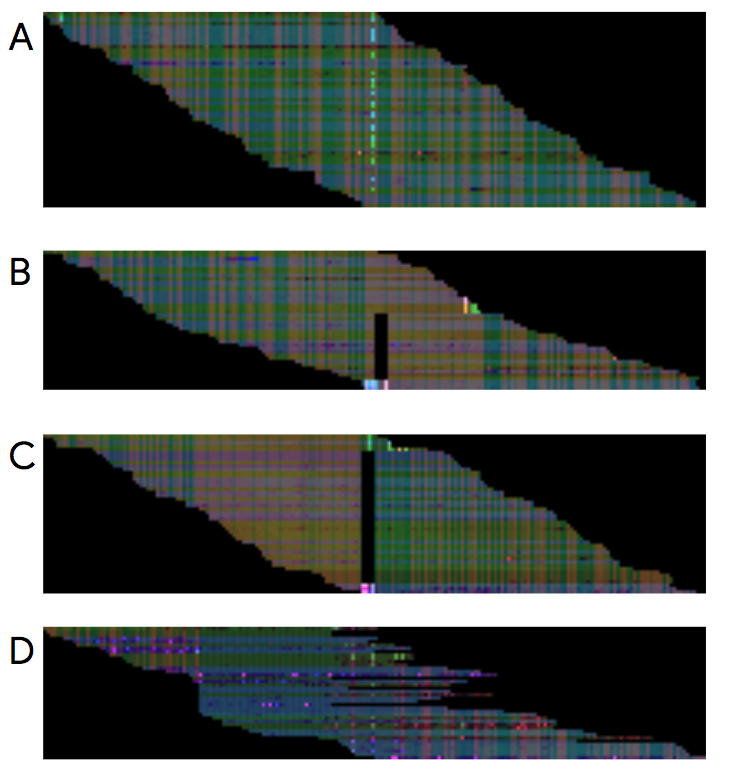
The make_example step uses the reference genome and the BAM file in order to produce the piled-up images needed for the prediction. To speed things up, the make_example step can be parallelised and take advantage of multiple machine’s cores.
Call_variants
The call_variants step performs the real variant calling: it uses the already trained prediction model to identify genomic variants.
Postprocess_variants
This step only converts the output of call variants in the standard and well-known VCF format.
A more detailed description of the steps can be found here.
DeepVariant in Nextflow
As shown in the following picture, the Nextflow version of the DeepVariant workflow contains not only the variant calling steps described above (darker blue ones) but also some preprocessing steps (light blue ones) as well.
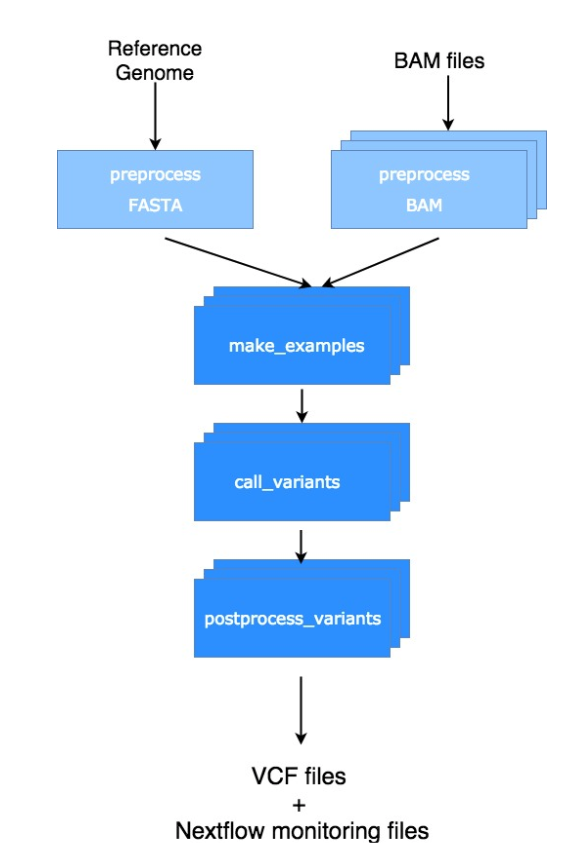
This allows for the whole analysis to run seamlessly and reproducibly instead of running manually 3 binaries with all the irreproducible mess, ineffectiveness and frustration that this may cause.
The workflow handles one reference genome and multiple BAM files as input. The variant calling for the several input BAM files will be processed completely independently and will produce independent VCF result files. The minimum set of input parameters is only composed by the version of the reference genome (available on Lifebit) and a folder where your bam files are stored. When running the pipeline in this way, the other needed files will be automatically created and the last release of google’s trained model for whole genome variant calling will be used.
It looks as simple as this:
nextflow run main.nf --hg19 --bam_folder path/to/folder/with/bam/files
Despite the possibility of keeping things this simple, complete control over all the configurations is ensured. Calling the pipeline can be as flexible as this:
nextflow run main.nf --fasta path/to/my/genome.fasta
--fai path/to/my/genome.fasta.fai
--fastagz path/to/my/genome.fasta.gz
--gzfai path/to/my/genome.fasta.gz.fai
--gzi path/to/my/genome.fasta.gz.gzi
--bam_folder path/to/folder/with/bam/files
--getBai true
--j 64
--modelFolder path/to/the/folder/with/my/model/files--modelName model.ckpt
It allows users to pass all the input files (compressed and indexed), define how many cores should be used for the parallelisation and even change the model which should be used. In this way, users can define their level of control.
Finally, the advantage of this approach is that the variant calling of the different BAM files can be parallelised internally by Nextflow and all cores of the machine are taken advantage of in order to get to the results faster.
DeepVariant at its best: Nextflow & Lifebit CloudOS
Running pipelines over the cloud can be a struggle and make you lose precious time if you are not an experienced cloud user: it begins with knowing how to fire up an instance and pick a suitable one, then how to transfer your data, how to clone your repositories, install all the dependencies and finally remembering to terminate the machines when the job is done to stay on budget. Honestly, this is not fun!! Which made us wonder in despair: why can’t the cloud be a simple compute tool that anyone could use without being cloud experts? We should not be losing time figuring out how the cloud works but rather concentrating on the results we are trying to obtain.
We built CloudOS to help relieve this pain – it saves time and helps you pick the fastest and most cost-effective configuration to run and scale your analysis over cloud.
It allows you, in a matter of a couple of clicks, to synchronise your cloud account as well as your GitHub or Bitbucket account. You can then run your pipeline on your data in a scalable manner over the cloud and still get some sleep at night!
CloudOS brings the easiness of running DeepVariant to a whole new level by providing a nice user interface which can guide the user through the journey of successfully and efficiently deploying and running variant calling analyses using DeepVariant over the cloud.
Epilogue on runtimes & costs
Using DeepVariant through CloudOS not only makes life easier, it actually speeds up running times and automatically makes the most out of the given resources.
We benchmarked DeepVariant running times over 10 different BAM files from UCSC on 3 different AWS machines with different resources (m4.16xlarge , cr1.8xlarge , c3.4xlarge).
We ran the binaries manually as described in the Google DeepVariant documentation taking care manually of preprocessing, transferring of data and cloud configurations (labelled “Bash Script” below) and DeepVariant as a Nextflow-based pipeline on CloudOS (labelled “Resource optimised with CloudOS” below).
Results show that DeepVariant in Nextflow on CloudOS outperforms the manual (bash script) version of DeepVariant across both time and costs. While looking at this results one should also take into account that, as a user, running DeepVariant on CloudOS took almost no time: you simply add input parameters and it’s done! On the other hand, running it manually needs time and effort, from generating all the needed input files (automatically handled in the Nextflow pipeline), manually moving data back and forth from Docker containers and having to trigger the computation multiple times in order to run DeepVariant on multiple BAM files. (NOTE: the time needed by these steps is NOT included in the benchmark, where only runtimes are shown). Not only are DeepVariant runtimes much lower by using CloudOS, but also the time needed for setup is eliminated.
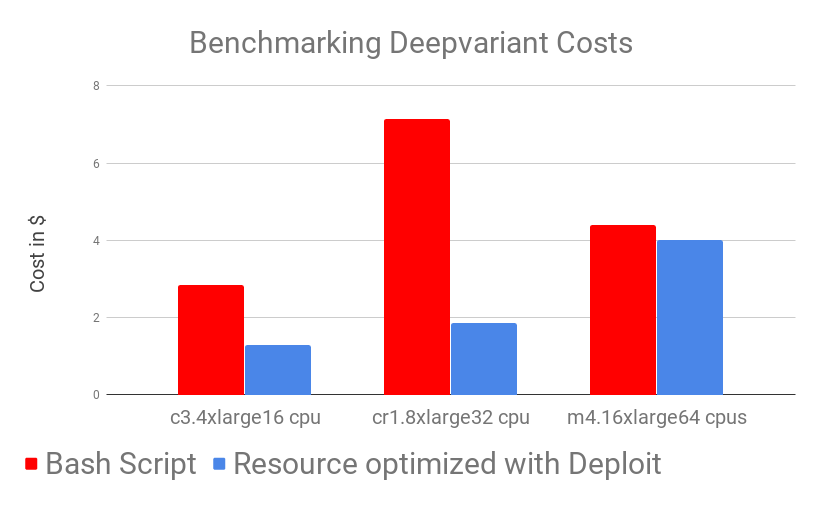
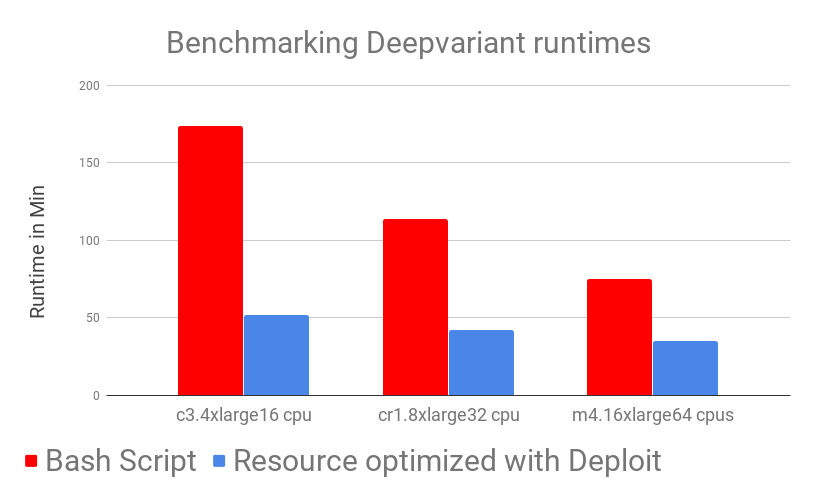
As can be observed in the benchmarking graphs, CloudOS can noticeably reduce time and lower costs. Time decrease is achieved by carefully using auto-scaling and parallelisation while low costs are ensured by CloudOS’ resource optimisation based on spot instances.
All data and material used for this benchmark can be found in this publicly available s3 bucket: s3://lifebit-deepvariant-benchmark-2018 . (The prices used for the benchmarking are based on the 16th June 2018 instances prices).
References
- Barkur S. Shastry, B. Jochimsen et al. SNP alleles in human disease and evolution. J Hum Genet. 2002;47(11):561.
- Bromberg Y. et al. Neutral and weakly non-neutral sequence variants may define individuality. Proc Natl Acad Sci U S A. 2013 Aug 27; 110(35): 14255–14260.
- Mathers J et al.The Biological Revolution: Understanding the Impact of SNPs on Diet-Cancer Interrelationships.The Journal of Nutrition 2007; 253S–258S.
We would like to know what you think! Please fill out the following form or contact us at hello@lifebit.ai. We welcome your comments and suggestions!
Featured news and events

2025-03-26 11:17:46

2025-03-14 15:45:18

2025-03-05 12:49:53

2025-02-27 10:00:00

2025-02-19 13:30:24

2025-02-11 08:39:49

2025-01-30 12:47:38

2025-01-28 08:00:00

2025-01-23 09:07:20

2025-01-08 13:58:41