.webp)
Genomics research is creating a big (data) problem
It’s no secret that the amount of genomics data generated is steadily increasing. By 2025, the compute resources needed to handle genomic data are expected to surpass those needed for both YouTube and Twitter combined (PLOS). And this extraordinary growth rate is not expected to slow down anytime soon.
With the advent of next generation sequencing (NGS), the price of sequencing a whole genome has gone down to about US$1,000 and is expected to go down even further as technologies continue to become more efficient and cost effective. This encourages the widespread use of sequencing, as we are seeing many new public and private initiatives based on sequencing technologies (i.e. Genomics England, Personal Genome Project, among others). What’s more, this technology is finally reaching the general public as millions of consumer genetic kits are being sold by companies such as 23andme and Ancestry, both of whom allow their customers to download their raw genetic data for personal use, or even for making a profit! As a direct consequence, we are undergoing a big data explosion, where the total amount of genomics data doubles approximately every seven months.
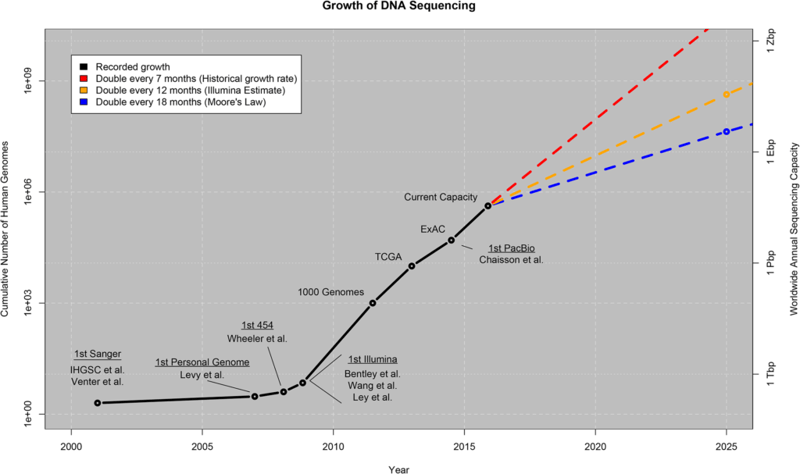
Producing genomic data is no longer an issue in today’s world. The bottleneck has now shifted to the analysis of the genomic data, where researchers are having a tough time keeping up with the throughput of the sequencing centres. As a result, genomic workflows (pipelines) are becoming more complex in order to deal with the amount of data and the ever-evolving complexity of analysis. But as workflows become more complex, we are also losing portability, reproducibility, and most importantly scalability.
Nextflow is a genomics workflow manager that solves these issues by basing its technology on the dataflow model and software containers. Containers, such as Docker and Singularity, are a standardised unit of software that wraps up the code and all its dependencies into a nice little package. Specifically, a container image will contain code, runtime, system tools, system libraries and settings, thereby allowing a quick and reliable run from one computing environment to another. This open source workflow manager has been adopted by a majority of the genomics sector, which highlights the need for a highly portable, reproducible and scalable workflow manager.
The major sticky point with open-source initiatives, however, is that standardisation is not always seen as a priority. This is a big issue, as different laboratories working with the same datasets might offer a range of different interpretations which is not ideal in any scenario. Automation that is inherent to Nextflow solves part of this problem by minimising human error and increasing reproducibility among different compute environments. However, by establishing best practices through requiring quality technical documentation, accreditation checklists and guidelines for workflow validation, reproducibility and traceability will naturally ensue.
The beginnings of a community of die-hard Nextflow fans
 Earlier in 2018, a new initiative, headed by Dr. Phil Ewels of NGI Stockholm, part of SciLifeLab in Sweden, was created and resulted in the nf-core community. Dr. Ewels describes how the project started:
Earlier in 2018, a new initiative, headed by Dr. Phil Ewels of NGI Stockholm, part of SciLifeLab in Sweden, was created and resulted in the nf-core community. Dr. Ewels describes how the project started:
“When looking at usage statistics and support requests for our NGI Nextflow genomics pipelines, it was clear that a lot of people were using our workflows as a starting point for new projects. We started working with other genomics labs such as QBiC in Tübingen and the ASTAR GIS in Singapore to collaborate on workflows and this grew into the nf-core project. The goal is to try to prevent the fragmentation of work – instead of everyone working with their own isolated copy of the same workflows, nf-core aims to bring the community together to work on a single set of gold-standard pipelines.”
The main goal of nf-core is to establish best practices concerning pipelines that are built with Nextflow. The main target audiences of nf-core are:
- Research facilities who benefit from highly automated and optimized Nextflow-based pipelines that generate reproducible analyses,
- Individuals who benefit by having access to portable and easy-to-use workflows,
- Developers that can have easy access to companion templates to check their code and facilitate repetitive tasks.
To date, there are a total of 14 available Nextflow-based pipelines in the nf-core repository, of which four have been released (eager, rnaseq, hlatyping, methylseq) and 10 others are still under development (deepvariant, ampliseq, smrnaseq, vipr, rnafusion, neutronstar, mag, chipseq, exoseq and lncpipe). You can check out the full list here. The nf-core community is made up of about 30 international contributors from many different institutions. Developing pipelines using nf-core tools and best practices better supports the bioinformatics community and allows everyone access to better quality pipelines.

What does it take for a Nextflow-based pipeline to make it to the nf-core released pipelines list, you ask? In order to assure highly-quality curated workflows, the nf-core community asks that in order to receive their seal of approval, a pipeline needs to fulfill certain requirements, some of which include:
- Has to be built using Nextflow, and have a MIT licence
- Software must be bundled with Docker and Singularity
- Continuous integration testing
- It should be a common pipeline and structure, with stable release tags
- The pipeline must be run with a single command
- Excellent documentation should be provided, along with GitHub repository keywords
- A contact person should be listed, and
- The pipeline cannot contain any errors in the nf-core lint tests (more about different error codes here)
It is important to note that since nf-core is still a new initiative, the recommendations and requirements are still evolving as the project gains a foothold in the Nextflow community.
As CloudOS is a platform for running Nextflow-based pipelines, we decided to contribute to this great community initiative by developing our DeepVariant pipeline under nf-core guidelines. Lifebit’s junior Bioinformatician, Phil Palmer, has recently gone through the process and details his experience in Nextflow Tutorial: Developing nf-core DeepVariant, a Google Variant Caller.
We would like to know what you think! Please fill out the following form or contact us at hello@lifebit.ai. We welcome your comments and suggestions!


